案例:通过深度测序分析乳腺癌中microRNA的序列和表达
背景: microRNAs调控很多对肿瘤发生非常重要的基因。乳腺癌中很多microRNAs是下调的,但系统的乳腺癌中microRNA的表达分析还没有被报道。
目的: 利用Illumina HiSeq2000测序平台对不同类型乳腺癌组织和细胞进行测序,分析其microRNAs表达差异。
结果:通过对11个正常乳腺组织,17个非转移组织,151个转移组织,及6种乳腺癌相关细胞系测序分析发现,正常组织中miR-21是高表达的,发生转移的乳腺癌病人的乳腺组织中mir-423是上调的,三阴乳腺癌病人中mir~17-92是特异性上调的,但是这些变化并不显著。这些结果表明,基于microRNAs水平对乳腺癌类型进行区分及预测转移统计学上可能是可行的,但是乳腺癌类型及转移的差异并不是由于富集的microRNAs的下调驱动的,提示在乳腺癌的发展过程中起作用的microRNAs不多。
原文索引:
Farazi TA1, Horlings HM, Ten Hoeve JJ(2011)MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer research, 71(13), 4443-4453
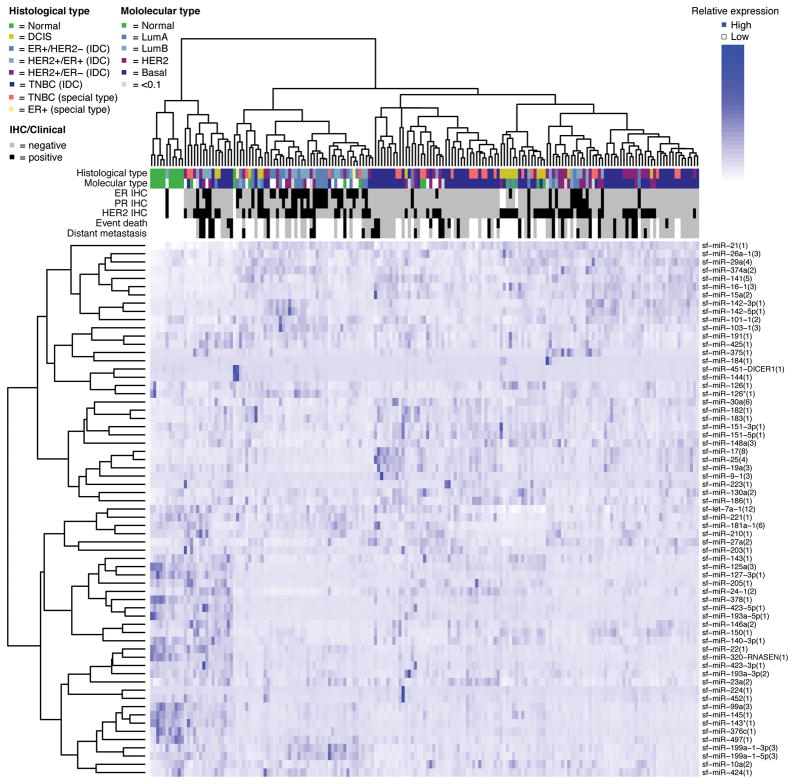
图1: 病人样品的microRNAs的聚类分析
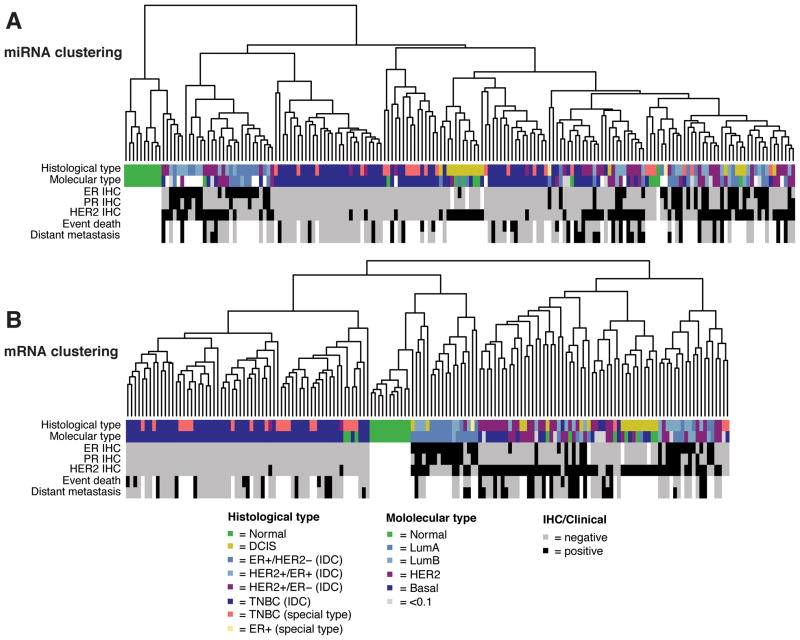
图2:microRNAs与mRNAs的聚类分析的比较